
Antibody-Antigen Complex Modeling
Yubeen Kim, Wooyeop Jeong, Chaok Seok* and Minkyung Baek*
AlphaFold-Multimer has limitations in antibody-antigen complex structure prediction, although AlphaFold2 predicts each monomer structure accurately. GalaxyTongDock, an ab initio docking tool, could be a solution for some problems when used with proper options.
Limitations of AlphaFold-Multimer in Antibody-Antigen Modeling
The main idea of Alphafold2 is to learn coevolution information from multiple sequence alignment(MSA). For antibody-antigen complexes, coevolution information is limited due to the unique generation mechanism of antibodies. As the antigen enters into our body, the antibody increases its affinity to the antigen rapidly through various mechanisms such as somatic hypermutation and VD(J) recombination. This antibody-specific mutation mechanism leads to lack of coevolution information between antibody and antigen.
Benchmark results for 81 antibody-antigen complexes shows that using unpaired MSA instead of paired MSA showed slightly better results, which means paired MSA information does not contribute much for antibody-antigen complexes. This is consistent with the fact that the antibody-antigen complex has limited coevolution information. Also, template information is helpful for high-quality model prediction. However, none of these methods produced decent performance.
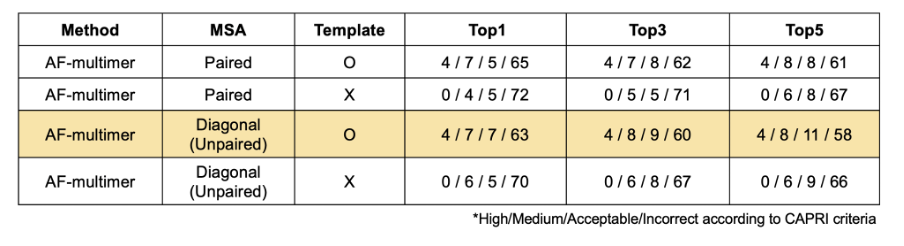
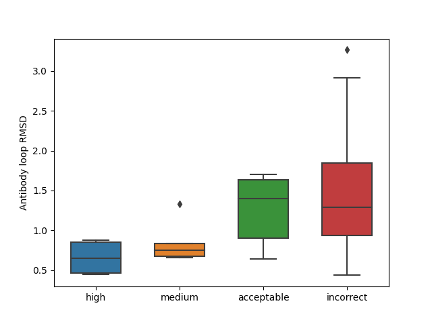
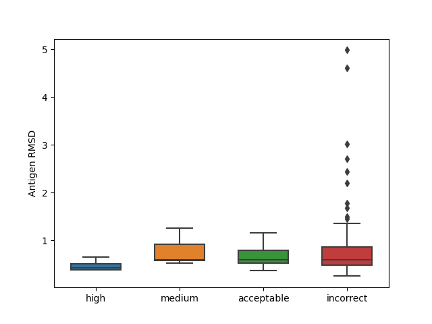
Further analysis showed that incorrect binding pose is the main reason for inaccurate antibody-antigen complex structure prediction. Antibody modeling does affect the quality of the entire complex, but overall RMSD value of antibody and antigen monomer is not very high. Also, as the wide range of antibody loop RMSD value in incorrect cases shows, high-quality monomers are not sufficient for high-quality complex modeling.
To determine whether the predicted structure has correct geometry or not, Predicted Aligned Error(PAE) score obtained by AF-multimer output can be used. For every pair (x, y) of residues in the structure, PAE is calculated by AlphaFold's estimate of position error at residue x, while the predicted and true structures are aligned on residue y. If the relative position of two domains is confidently predicted, the PAE values will be low (less than 5Å) for pairs of residues from each domain.
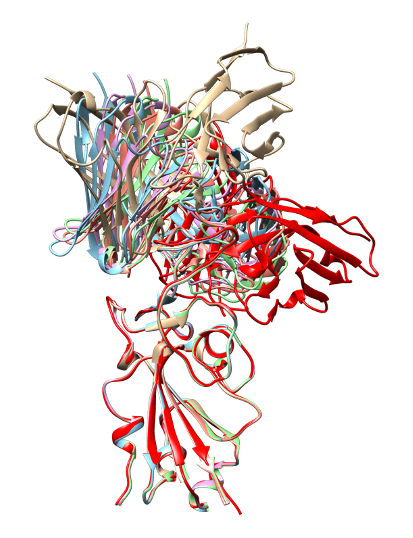
For example, in the case of 6yx9, which has correct predicted structure, has low PAE score for all pairs of monomers for model ranked #1 to ranked #4. However, for inaccurate predicted ranked #5 model(yellow), PAE score between antibody and antigen residue is high.
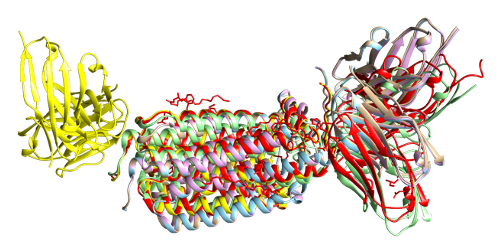
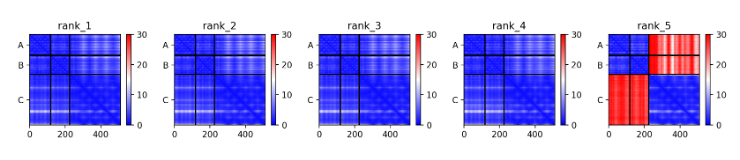
Also, in the case of 7BEM, all predicted structures show inaccurate results, and the PAE score between antibody and antigen is relatively high compared to intra-antibody PAE score.
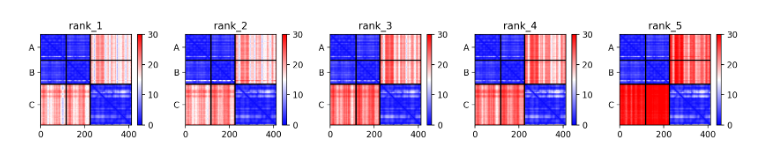
This implies that PAE score could discriminate between correct and incorrect binding pose. For structures that are not expected to be well predicted, other methods such as ab initio docking can be used.
More accurate prediction with GalaxyTongDock
As mentioned above, users could fail to get accurate predictions for antibody-antigen complexes with AlphaFold-Multimer. In this case, users can use the GalaxyTongDock1, ab initio docking tool in addition to AlphaFold2. GalaxyTongDock based on 3D FFT algorithm, returns the predicted complex structure with the least energy using parameters optimized from ZDOCK. Prediction of the program is mainly affected by shape complementarity of the backbone structure, so given crystal structures or accurately predicted structures, it could predict highly accurate complex structure.
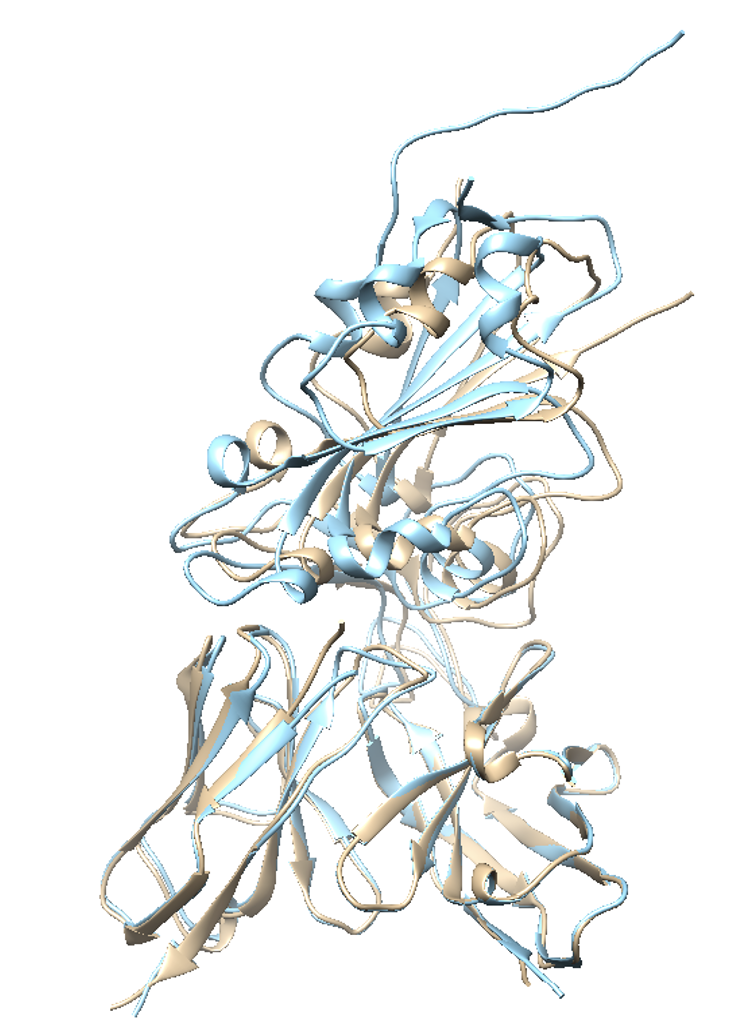
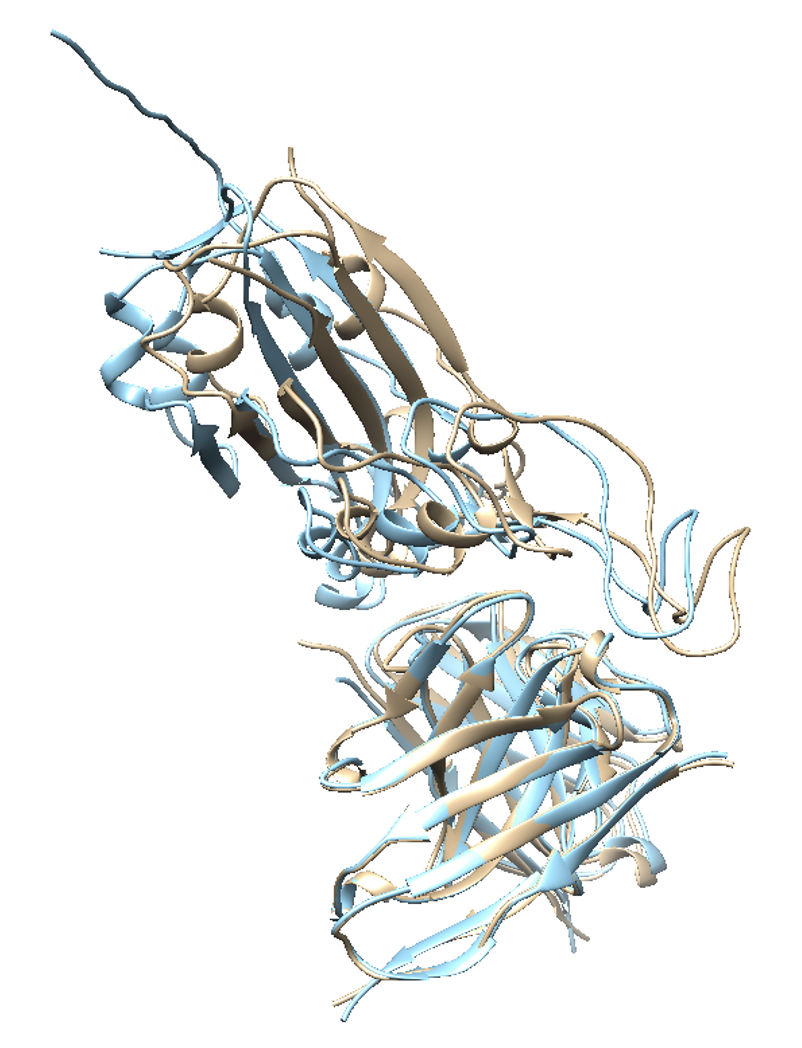
As the predicted structure of 7BEM implies, if the complex has large buried surface area (BSA) and good shape complementarity (due to loops in the example), GalaxyTongDock predicts the structure accurately because its energy score is favorable with the highly complementary interface. However, it implies that we have to be careful with targets with small BSA and low shape complementarity. Then, how can we get more reliable predictions for target complexes with a flat interface and small BSA?
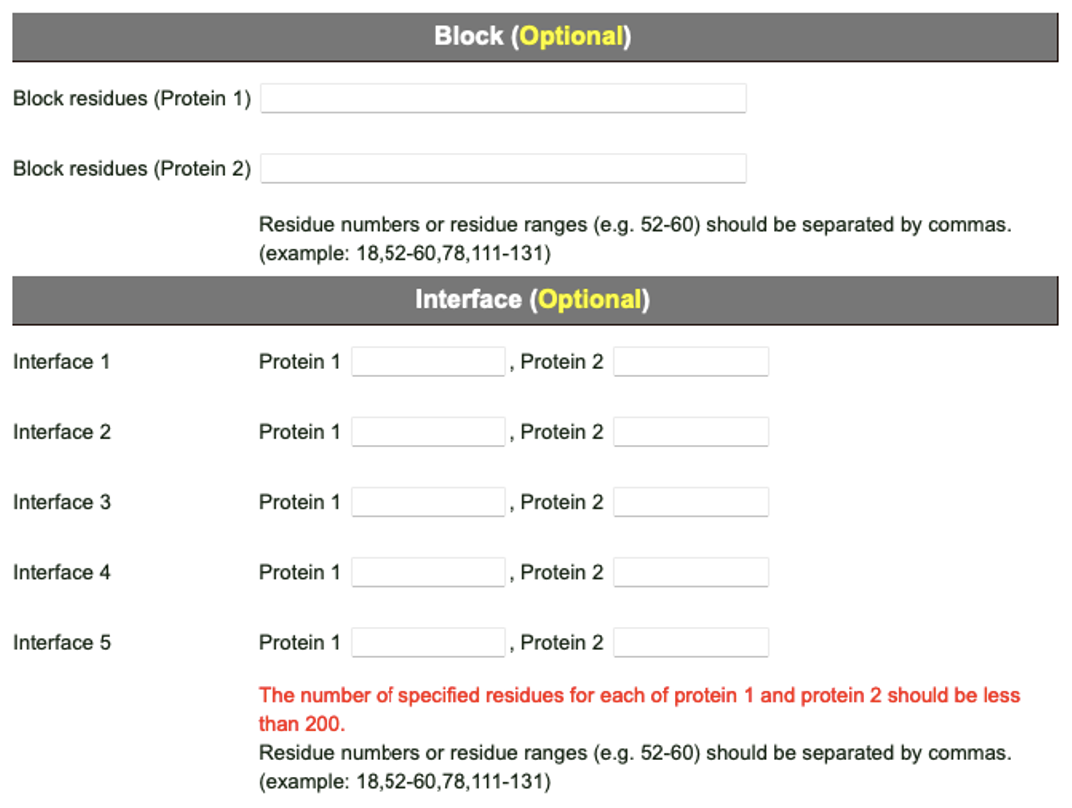
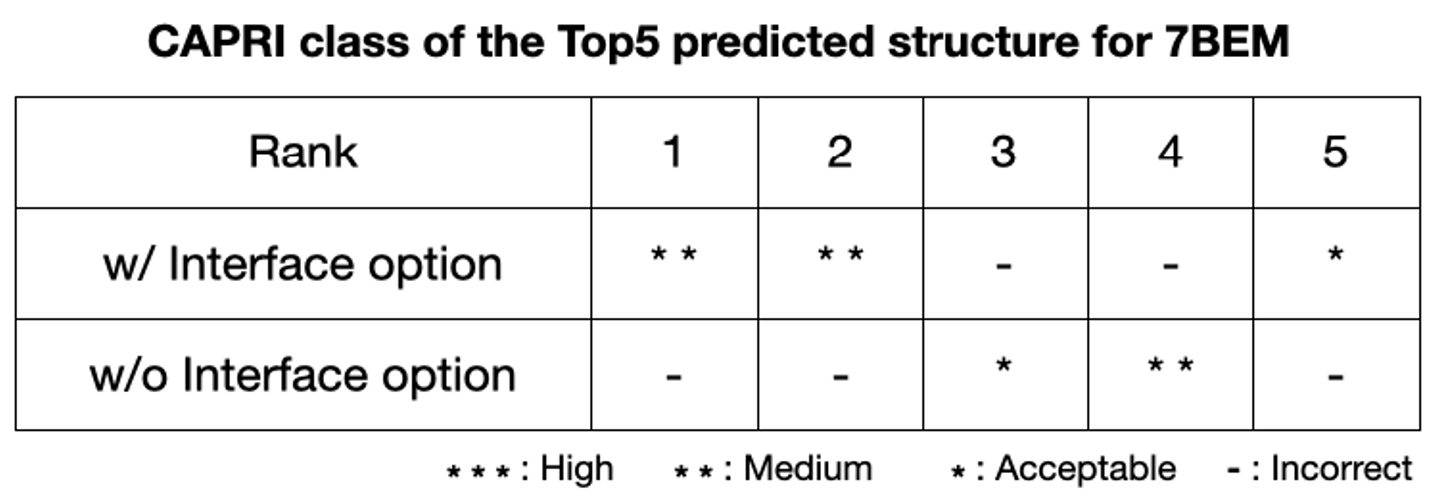
GalaxyTongDock provides “Block” and “Interface” options as a solution for the problem above when some clues are available for the interface. These options add high penalties for blocked residues and advantages for interface residues when they are at interface. Given the interface option to the 7BEM, the 1st ranked model is of medium quality (ranked 7th without interface option), and the ratio of the hit prediction (above acceptable in CAPRI criteria) becomes higher. GalaxyTongDock would be more powerful when these option are combined with experimental data such as metagenesis data or target epitope information.