
Integrative Structure Modeling
Seho Lee, Chaok Seok* and Hahnbeom Park*
For large biomolecular complexes, experimental structures for composing units and in silico complex modeling could be combined to unveil their behavior. Deep-learning based modeling such as AlphaFold-multimer and an ab initio docking tool such as GalaxyTongDock could help researchers to predict structures corresponding to different states.
Introduction
Integrative structure modeling would require combining computational resources and experimental analysis and information. Due to the development of deep-learning-based protein structure prediction, it is possible to predict the complex structure, enabling the experimental scientists to experiment more efficiently before starting the investigation. Conversely, by referring to the results of experiments as clues in calculations, a complex structure that experiments have not determined can be made into a more convincing structure through calculations.
Integrative modeling is generally required in large biomolecules. It means that integrative modeling involves a lot of memory and computational costs. Therefore, a proper trade-off between the size of the biomolecule and the computational cost is required without compromising accuracy for acceptable computing cost and time. In this case, the experimentally known properties of an objective biomolecule are used to avoid excluding important and characteristic parts of the study object from the calculation. In addition, proteins with similar functions, information on typical amino acids, and researchers' intuition can all help extract necessary structures.
AlphaFold2 and AlphaFold-multimer are powerful protein structure prediction tools that apply to both tasks mentioned above. In particular, Alphafold-multimer is a powerful and valuable tool for multimer prediction as it has a success rate of about 75% on the benchmark (better than "acceptable" based on CAPRI criteria) in flexible protein interfaces.
Despite the high prediction accuracy of AlphaFold-multimer, this may indicate that the researcher was not satisfied. In this case, you can try docking using the information that the researcher wants. GalaxyTongDock is a 3D FFT-based protein-protein docking method that uses algorithms such as ZDOCK. If researchers specify amino acids between two proteins in contact, it expects the least energy complex structure using energy parameters optimized from ZDOCK. If the starting protein structure is not accurate, or if the researcher does not have any available protein structure, the protein structure can be generated using AlphaFold2. Currently, AlphaFold2 can be run with ColabFold efficiently.
Track 1
- Prepare sequence of protein-protein complexes
- Remove unnecessary parts of sequences.
- Run AlphaFold-multimer! (Using ColabFold) (total <2000AA recommended)
- For multimer, prepare sequences like (seq1) : (seq2)
e.g.
ACDEFHIKLM:NPQRSTVWXYZ - Select MSA option (paired MSA recommended)
- For multimer, prepare sequences like (seq1) : (seq2)
e.g.
- Superpose resulting structure with corresponding mono-structures.
Track 2
- Prepare sequence of protein-protein complexes
- Prepare 3D structure of each chain from PDB bank or search from Alphafold2 structure.(Uniport search, or EBI AlphaFold Database search)
- If there is no available structure, run alphafold2 for each single chain.
- Remove unnecessary parts of sequences.(total <2000AA recommended)
- Assign binding residues(GalaxyTongDock input preparation)
- Assign residues to be blocked
- Run GalaxyTongDock with 4,5
- Superpose the AlphaFold structure of unused parts of sequences.
Examples
Expected Mechanism of ECM29 - proteasome complex disassembly
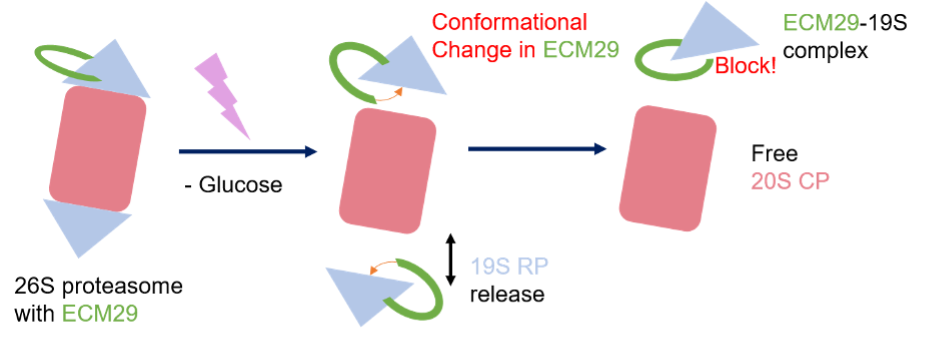
Protein-protein complex
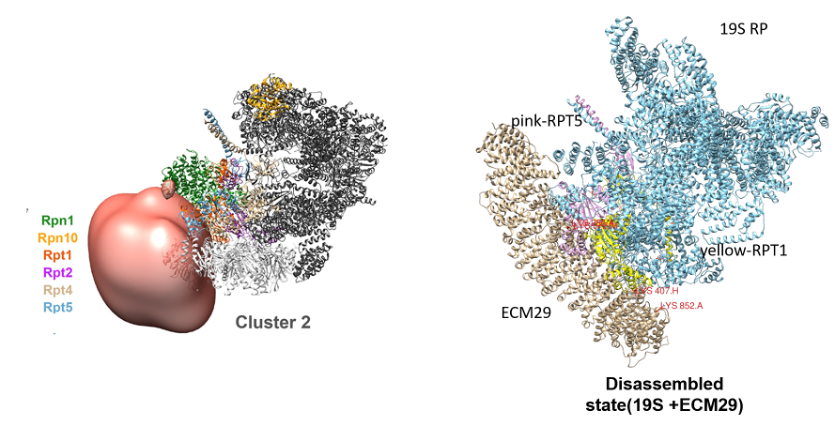
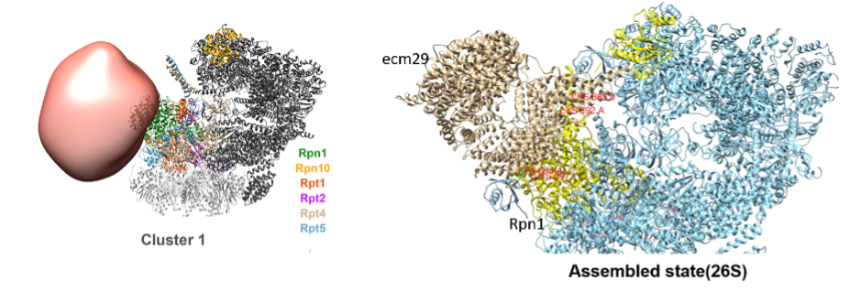
Overall Structure of ECM29(ECPAS)-Proteasome complex in Assembled, Disassembled state.
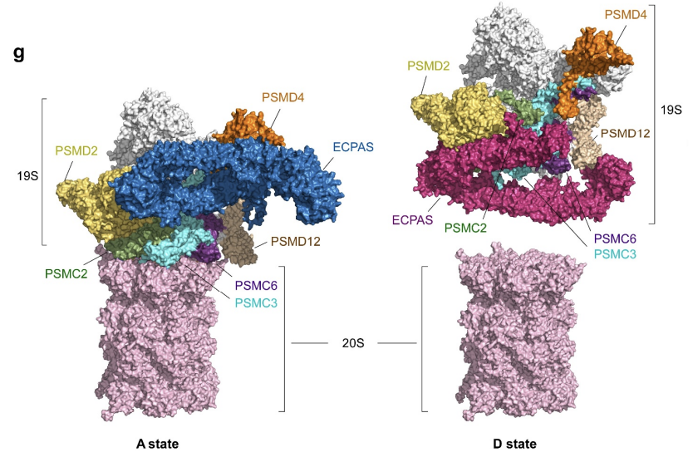
References
- YIN, Rui et al. Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Science 2022, 31 (8), e4379.
- Wang, X.; Chemmama, I. E.; Yu, C.; Huszagh, A.; Xu, Y.; Viner, R.; Block, S. A.; Cimermancic, P.; Rychnovsky, S. D.; Ye, Y.; Sali, A.; Huang, L. The proteasome-interacting Ecm29 protein disassembles the 26S proteasome in response to oxidative stress. The Journal of Biological Chemistry 2017, 292 (39), 16310-16320. DOI
- Choi, Won Hoon, et al. ECPAS/Ecm29-Mediated 26S Proteasome Disassembly Is an Adaptive Response to Glucose Starvation. bioRxiv 2022. DOI
- Park, T; Baek, M; Lee, H; Seok, C. GalaxyTongDock: Symmetric and asymmetric ab initio protein-protein docking web server with improved energy parameters. J Comput Chem. 2019, 40 (27), 2413-2417. DOI